
X射线吸收精细结构——原理及应用
摘要
X射线吸收精细结构(X-ray Absorption Fine Structure,XAFS)是一种基于同步辐射X射线源的光谱分析方法,可进一步分为X射线近边吸收精细结构(XANES)和扩展X射线吸收精细结构(EXAFS)。XAFS在近十几年来迅速发展,并成为表征材料结构的常规方法。本文介绍XAFS方法的发展历程、基本原理以及其在纳米电催化材料结构表征中的应用。
关键词: X射线吸收精细结构,光谱学,材料科学
Abstract
X-ray Absorption Fine Structure (XAFS) is a spectroscopic analysis method based on synchrotron X-ray sources, which can be further divided into X-ray Near Edge Absorption Fine Structure (XANES) and Extended X-ray Absorption Fine Structure (EXAFS) according to the energy region of the spectra. XAFS has developed rapidly in the past decades and has become a routine method for characterizing the structure of materials. This paper introduces the development of the XAFS method, its basic principles, and its application in the structural characterization of nanomaterials.
Keywords: X-ray absorption fine structure, spectroscopy, materials science
第一章 XAFS的发展历史
1.1 X射线的发现及X射线谱学的早期发展
1901年,伦琴因发现X射线获得了首届诺贝尔物理学奖。X射线发现之后,伦琴、波赫和科尔等科学家便开始探究X射线的性质。
马克思·冯·劳厄(Laue)、Friedrich和Knipping通过实验阐明了X射线的性质。当Friedrich和Knipping用X射线照射硫酸铜晶体时,照相底片上出现了斑点,这无疑是因为入射的X射线在晶体中发生了衍射。劳厄进一步建立了数学模型以模拟X射线在三维晶格中的衍射,从而揭示了晶体的结构。1914年,劳厄因发现X射线晶体衍射现象获得诺贝尔物理学奖。
劳厄的工作开创了X射线研究的两个分支:晶体结构测定与X射线光谱学。威廉·劳伦斯·布拉格(W. L. Bragg)沿着前者前进:小布拉格受到衍射图样的启发,使用他父亲的光谱仪研究了氯化钠和钾、金刚石、方解石、萤石、铁黄铁矿、萘和蒽等物质的结构,发现氯化钠并非以分子形式存在,这在当时的化学界是一个颠覆性的认知。而小布拉格的父亲,威廉·亨利·布拉格(W. H. Bragg)致力于X射线光谱学的研究,并证实了X射线管光谱与了查尔斯·巴克拉提出的K线和L线相匹配。布拉格父子因他们的开创性工作于1915年获得诺贝尔物理学奖。
1.2 XAFS的发展历程
1.2.1 XAS的早期发展
事实上,早在19世纪20年代,德布罗意、Fricke和Hertz等人就已观测到X射线吸收边,但X射线吸收光谱(XAS)的广泛应用要比X射线衍射法(XRD)滞后很长时间。限制XAS谱学发展的主要因素有二:一是当时的研究集中于XRD上,使得XAS相对不受重视,二是当时X射线仪器条件较差(强度低,单色性差)。
XRD极大地推进了固态物质的研究,这是因为其可以确定有序结构,但当这种有序结构不存在时(如在非晶态材料、高度无序的系统以及溶液体系中)XRD就无能为力了。XAS虽然可以弥补XRD的缺点,但XAS谱学是在20世纪80年代强X射线光源(如同步辐射)技术成熟以及计算机技术发达之后才真正流行起来的。在此之前,科学家们进行了许多有益的尝试,其中有两位科学家Kossel和Kronig对XAS的谱图解析进行了探索。1920年,Kossel指出XAS精细结构的出现与吸收原子内层电子的激发相关,但难以指认谱峰归属;1931年,Kronig将XAS的振荡吸收峰归结于长程有序的结构,但实验中观察到的振荡现象与理论偏差较大,以致在XAS现象发现后的前50年,科学家们一直没能对XAS谱图形成统一的认识。
1.2.2 同步辐射光源的出现及XAFS的进一步发展
1954年,Johnston和Tomboulian等人首次运用粒子加速器释放的同步辐射(SR)测定了硼的K边吸收光谱,这标志着同步辐射技术应用的开端,但由于早期同步辐射光源不稳定以及设备操作复杂、费用高昂等原因,其真正广泛应用于XAS是在10~15年后电子储存环的出现之后。电子储存环发射的同步辐射更加稳定可控,使得其更适用于科学研究;同时,同步辐射光源高准直性、高偏振性和高强度等特点大大提升了XAS谱图的质量,使得研究者能方便地获得高分辨率XAS谱图从而观察其精细结构。总的来说,同步辐射使XAS进化为XAFS,并进一步将XAFS的研究划分为两个部分:X射线近边吸收精细结构(XANES)和扩展X射线吸收精细结构(EXAFS)。
XAFS理论也随着仪器设备的进步突飞猛进。1971年,Sayers、Stern和Lytle的研究证明XAFS谱图的振荡是光电子干涉造成的,并且可以从振荡谱图中提取出局域结构信息。这是对30年代XAS早期理论的重大突破。1975年,Lee等人将多重散射(Multiple Scattering)理论引入EXAFS,这同时也代表XANES谱图计算研究的开端。此后,XAFS的理论不断完善。其中,EXAFS的发展速度快于XANES,这是由于XANES谱图较为复杂,难以计算导致的。
至今,XAFS技术已经成为材料科学研究中不可或缺的表征手段。EXAFS,提供了大量基于单散射过程的键长信息,而XANES不仅能够提供电子态数据,还能基于多重散射过程给出局部配位结构。
第二章 XAFS的基本原理[1-2]
2.1 XAFS的产生原理
2.1.1 电子的激发、跃迁与电离
当X射线照射原子时,如果入射X射线的能量与原子内层电子能量相近,内层电子在吸收X射线光子后被激发,产生跃迁(从某一组态跃迁至更高能量的另一组态)或被电离(如果X射线的能量足够强)成为光电子。使电子电离所需要的能量称为吸收阈能。X射线吸收系数在吸收阈能附近突然增大,随后随着X射线能量继续升高,X射线仍会被吸收,而电离出的光电子动能随之增加,在光谱图上体现为产生吸收边(edge)。
电子从内层轨道向外层空轨道的跃迁需要满足以下几个条件。
(1)外层要有未被占领的空轨道。
(2)服从配位场理论和光谱跃迁选律的规则,因此可以由光谱数据获取关于配位环境和局域对称性的信息。
当原子的K层电子被激发,产生的吸收边称为K边,当原子的L、M等层被激发时,由于电子组态(如ns,np1/2,np2/3)的不同,可进一步分为LI边、LII边、LIII边等吸收边。同一元素K吸收边的阈能一定大于L边。
2.1.2 散射和干涉
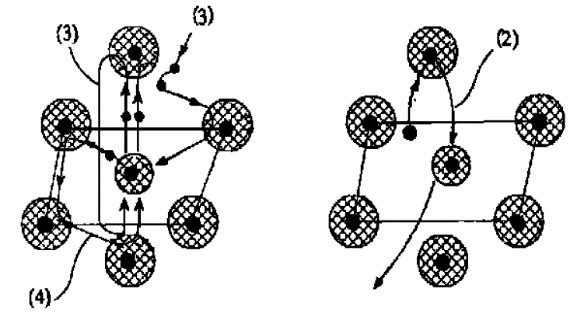
光电子的动能随入射X射线的能量不同而有所不同,如果光电子动能较大,则光电子一般只会被近邻原子单次散射后逸出,这也是后续讲述的EXAFS方法的基础;当光电子动能较小时,则会被不止一个近邻原子多次散射,即发生多重散射。两种散射的示意图见下。
2.2 XAFS谱图的获取与处理
2.2.1 XAFS光源
XAFS所使用的X射线源不仅要求具有高强度,还要求具备高的单色性。主要原因有二:一是由于不同组态的电子跃迁所需能量相近,因此测量吸收边前和边后部分时,需要很高的分辨率(0.1eV),才有可能辨别出不同组态的跃迁吸收;二是因为EXAFS谱图的振荡部分(EXAFS函数,见2.4节)进行傅里叶变换时,需要尽可能多的数据点才能保证变换的精度和数据可靠性。因此,一般利用同步辐射光源进行XAFS的测定。
2.2.2 XAFS谱图的分区
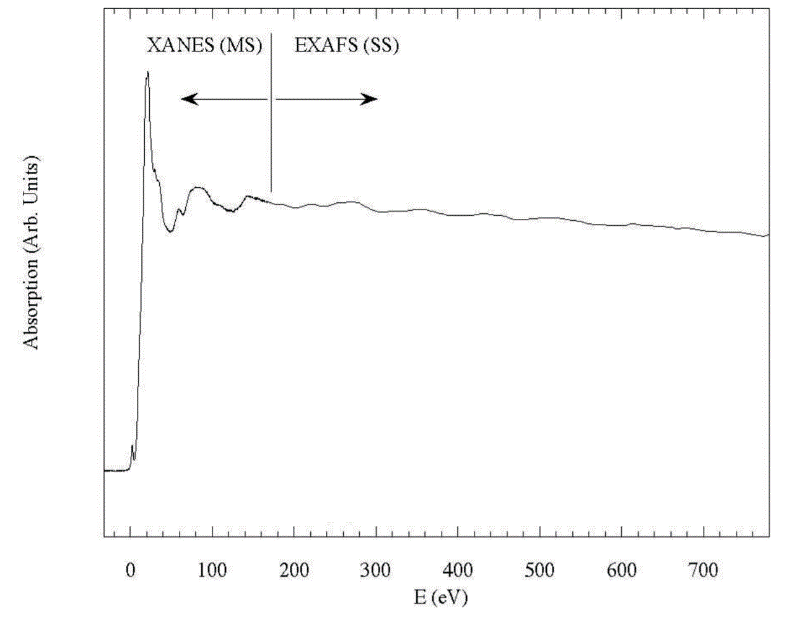
按物理机制分类,XAFS谱图可以分为三区:
(1)中心原子内层电子跃迁主导的边前(pre-edge)及吸收边;
(2)中心原子受激发的光电子被近邻原子多重散射主导的形状共振(shape resonance)区(10-70eV),(1)区和(2)区常被归为XANES区;
(3)中心原子受激发的光电子被近邻原子单散射主导的EXAFS区(50-1000eV)。
2.2.3 XAFS谱图的预处理
(1)能量校正。对于一组样品中同一元素的XAFS测定,应当选取与待测样品同元素的稳定化合物作为标样,取起吸收边上最大值为参考点,在测定前后测定该点能量位置以证明没有移动。
(2)归一化。归一化的目的在于让XAFS(尤其是XANES)上各精细结构在统一的尺度上显示出来。进行归一化首先要确定能量零点。一般对XANES谱边前20eV至边后70eV求导,取导数极大值处为能量的相对零点。接下来,以吸收边台阶的高度为1进行能量归一化。
(3)背景扣除。XANES谱图是叠加在正常吸收边上的,因此应当扣除背景。扣除背景之后的XAFS数据才能用于计算。不过,作图时,可只画出归一化但未扣除背景的谱图。
2.3 XANES谱图解析
现在对XANES谱图的解析主要集中在边前峰上,而对于多重散射造成的形状共振理论尚未成熟,这一部分的谱图识别主要是通过标样对照。
XANES中的边前峰可以给出配位结构信息,而吸收边对应的吸收阈值可反映元素的氧化态(见应用部分)。
现以Mo的氧化物及多酸化合物为例[3]对XANES的谱图解析进行简单介绍。
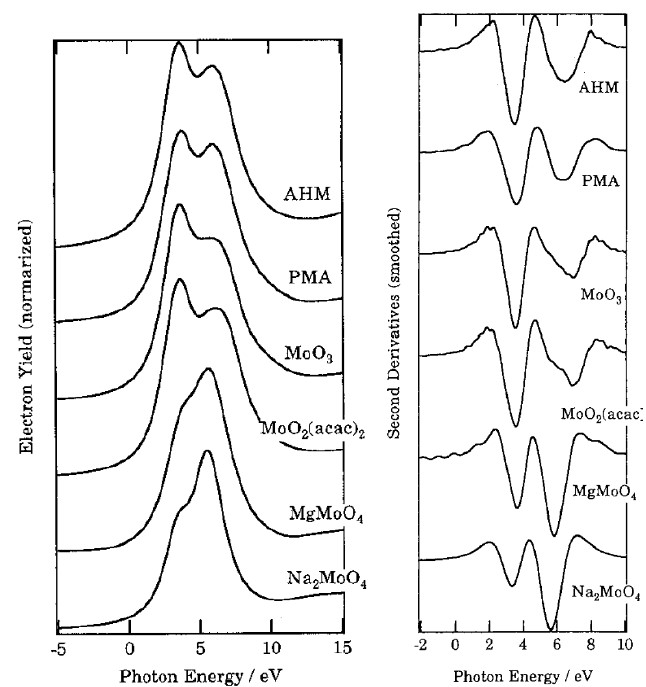
含Mo6+化合物的L3边主要由2p3/2至4d的跃迁贡献。从图3中可看出,虽然Mo的价态均为6价,但谱图出峰有所不同。虽然所有的样品都出现了两个峰,但MgMoO4和Na2MoO4的谱峰在高能量处的峰较强,而其他四个样品在低能量的峰较强。这是因为MgMoO4和Na2MoO4具有[MoO4]四面体结构,XANES谱图中两个峰分别对应4d轨道分裂的t2(dxy,dyz和dxz)能级和e(dx2-y2和dz2)能级,强度比为3:2;其他四个样品具有[MoO6]八面体结构的吸收峰对应4d轨道分裂的t2g能级(dxy,dyz和dxz)和eg(dx2-y2和dz2)能级,强度比为2:3。
除了可以用XANES观测到配位环境的不同,还可以根据求导后的谱图数据进一步计算各种晶体场的分裂能。由二阶导数图得知,[MoO4]四面体场的分裂能约为2.2-2.3eV,而[MoO6]八面体场的分裂能约为3.3eV。
2.4 EXAFS谱图解析
EXAFS谱图解析的核心在于对振荡曲线(EXAFS函数)进行傅里叶变换获得结构参数。
EXAFS函数通常用参量表示,有
它表示X射线吸收系数μ归一化后的振荡部分,其中k为出射光电子的波矢:,为光电子波长,代表孤立原子的吸收系数,而代表扣除背底后吸收边高能侧的吸收系数。
与以下几个因素有关:
(1)与吸收原子周围的第j近邻原子壳层中的同种原子数Nj,距离Rj及原子散射因数fj(2k)有关,这一部分可表示为:
(2)与散射光电子的位相改变有关。位相改变分为两部分:一是光程差引起的位相差以及出射和散射引起的相移。这一部分可表示为:
(3)与第j壳层原子的漫散分布程度有关。它包含热振动和原子无序分布的影响。设σj为对Rj的均方根偏离,则该影响可表示为Debye-Waller温度因数型的项:
(4)与出射光电子保持原状态的传播距离有关,该因子为
综合以上因素,可得到表达式:
将EXAFS谱图进行背景扣除和归一化后,按照下式
将光电子的能量转换为波矢k,再求出,接着进行傅里叶变换,得到径向分布函数Φ( R ),该径向分布函数反映了在距离中心原子R距离处出现原子的概率,Φ( R )具有极大值的R值可近似为各配位层的配位半径。
第三章 XAFS的应用
3.1 XAFS谱图信息汇总
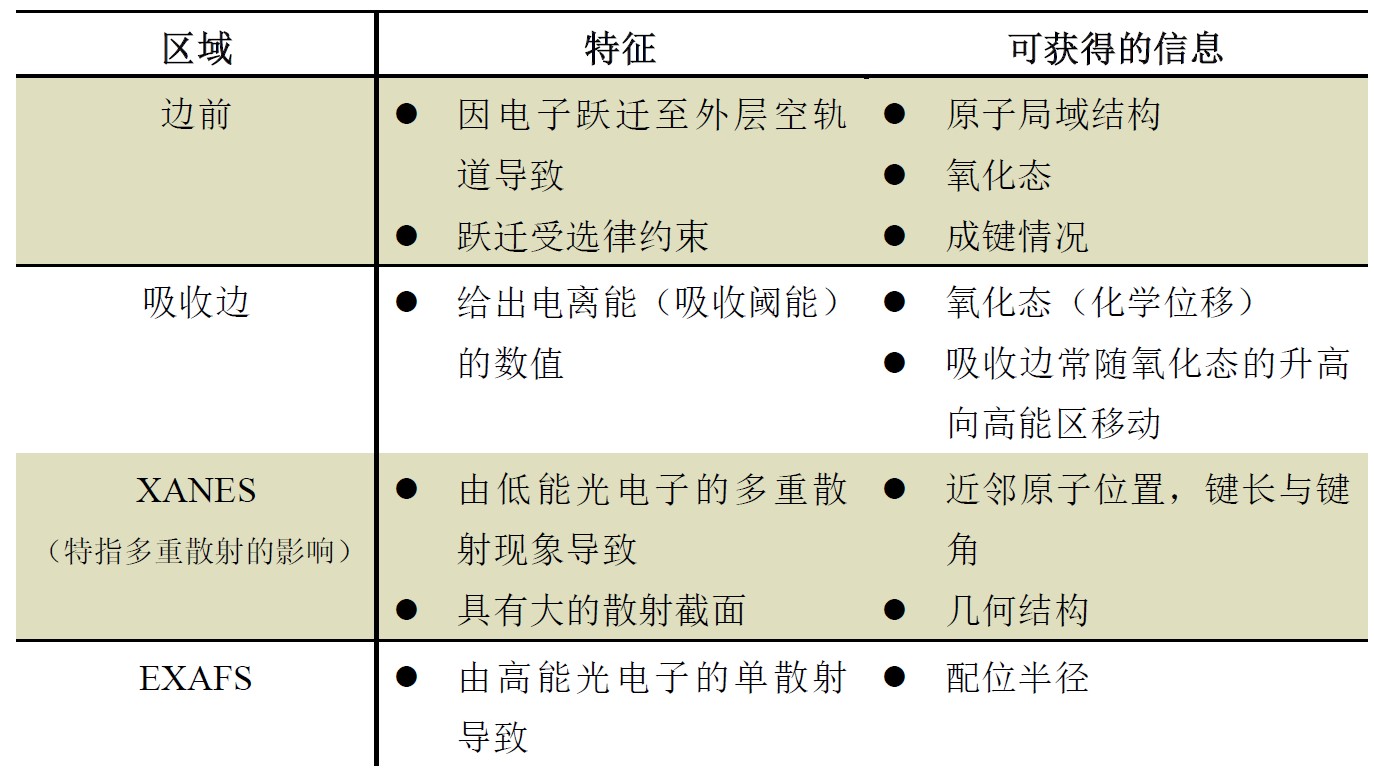
根据上表总结,从XAFS谱图的不同区域可获取关于材料结构的许多信息,以下,在纳米催化材料领域,举几个例子介绍之。
3.2 XAFS的应用实例
3.2.1 元素分布分析
从EXAFS可以获得原子的配位半径,因此EXAFS可应用于元素的分布分析。如当金属元素呈单原子分散时,由于两个相同原子的距离很远,M-M键对应的峰将明显弱于元素呈聚集态时的M-M峰,见下例。
例1[4]:研究者通过将少量的Mo元素掺杂至La2CoMnO6双钙钛矿结构中,其Mo K边 EXAFS谱图经FT变换后的径向分布函数(图4)显示只存在键长1.2Å的Mo-O键而不存在键长3.1-3.3Å的Mo-Mo键(相对于MoO2和MoO3而言),这说明Mo物种在双钙钛矿结构中达到了原子级分散。
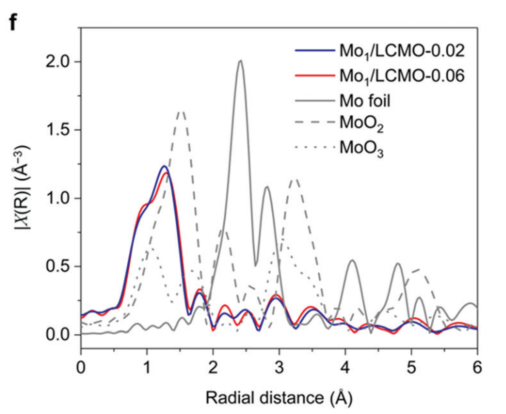
3.2.2 成键情况分析
例2[5]:钙钛矿电催化材料中,金属-氧键的共价性是影响催化剂活性的重要因素。Jin Suntivich等人通过O K边XAFS数据(图5a)得出M-O共价性随3d电子数目增多而增大的趋势。他们将O 2p光谱边前峰面积的大小作为衡量M-O键共价性的指标,并通过计算峰面积得到d电子与M-O键共价性的线性关系(图5b)。
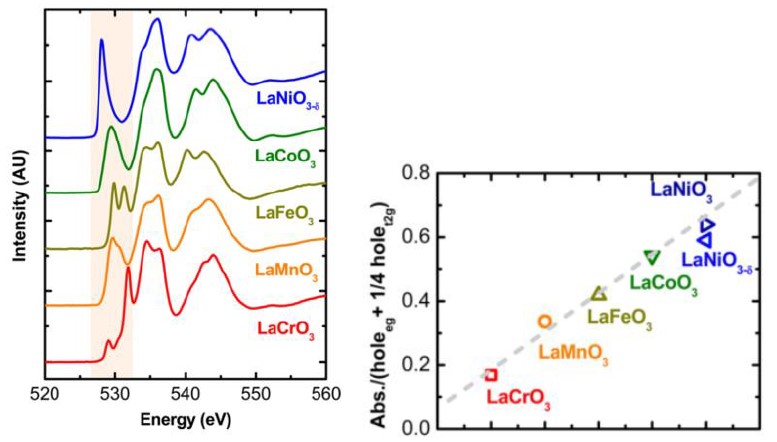
3.2.3 元素价态与配位环境分析
通过XAFS进行元素价态和配位环境的分析,主要从XANES的吸收边和EXAFS傅里叶变换数据入手。以下举几个例子。
在前述例1中,从 Mo K边XANES谱图(图6)中可以看出,Mo掺杂后的双钙钛矿材料Mo/LCMO,其吸收边十分接近于MoO3,说明Mo在材料中的氧化态接近+6.
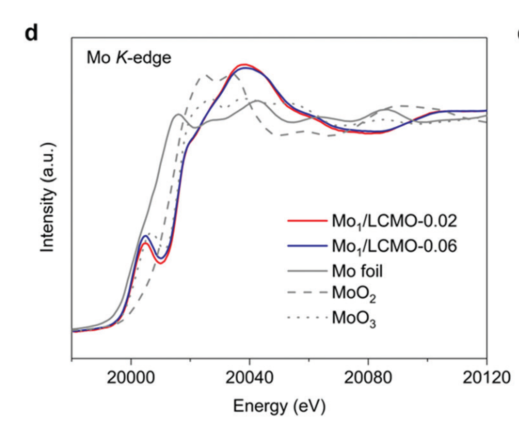
研究者还研究了Mo的掺杂对钙钛矿中Co和Mn价态的影响。从Co和Mn的XANES谱图(图7)中可看出,由于Mo/LCMO和LCMO的吸收边几乎重合,因此Mo的掺杂没有对Co的价态产生影响;而对于Mn的吸收边,Mo/LCMO稍向低能态移动,说明Mn的氧化态出现少许降低(出现了少许Mn3+)。高自旋的Mn3+是有利于ORR反应的,研究者们通过XAFS谱图很好地阐明了Mo/LCMO的催化活性来源。
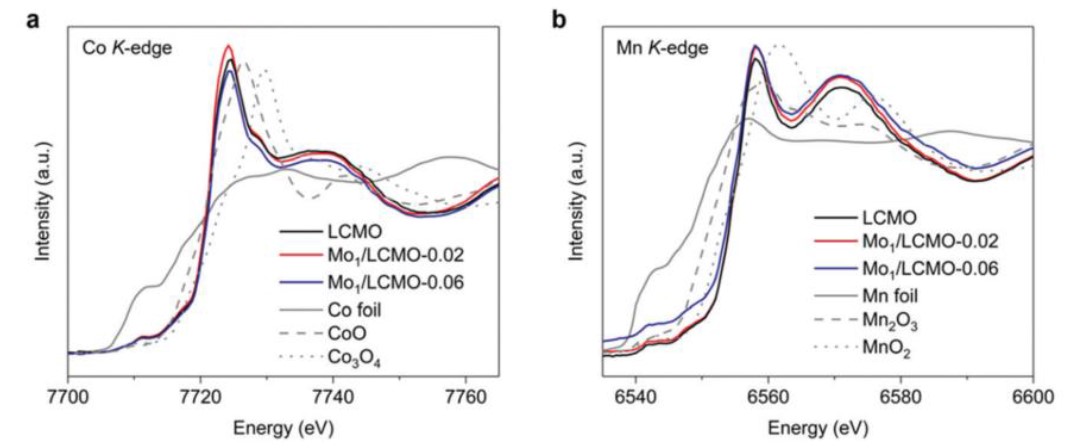
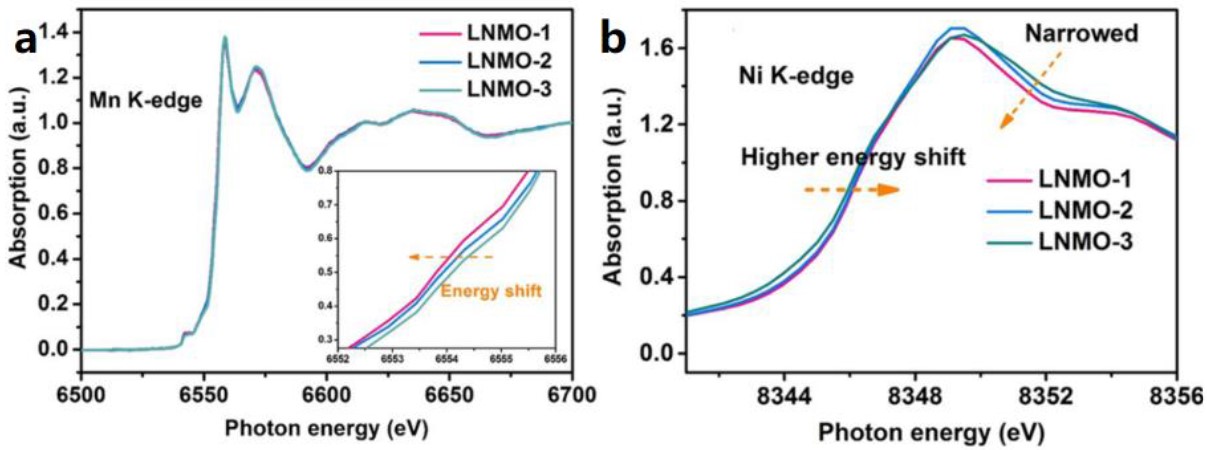
由于La XPS峰经常与Ni XPS峰强烈重叠,通过LNMO中的XPS分析直接推断Ni的价态变化并不可靠。而XANES分析可以完美地解决此问题,详见下例。
例3[6]:研究者在不同的温度下合成La2NiMnO6材料,并研究了Ni3+-O-Mn3+的振动超交换作用。通过Mn和Ni的K吸收边XANES谱图(图8)可发现,随着煅烧温度的提升,Mn的吸收边向低能区位移(价态降低),而Ni的吸收边向高能区位移(价态升高),这说明随着温度的升高,Mn4+与Ni2+通过氧桥连接的振动超交换作用就越显著,二者的氧化态和eg轨道电子填充状态趋于统一。
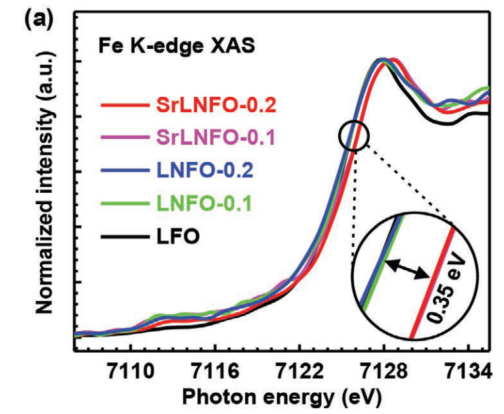
例4[7]:Gaoliang Fu等人将Fe掺杂进LaNiO3钙钛矿结构,并通过Fe K边XANES谱图(图9)对比吸收边能量,发现LaNixFe1-xO3和SryLa1-yNixFe1-x材料中,Fe的吸收边向高能区位移,说明有Fe4+的生成。后续的DFT计算表明由于Fe4+诱导使得Ni3+/Ni4+的转变更为容易,降低了OER决速步的反应能垒,进而提升催化性能。
例5[8]:Xiao Liang等人通过合金化策略设计出了一种SrZrO3(SZO)与SrIrO3(SIO)的钙钛矿型固溶体材料SZIO。对SZIO、SZO、IrO2和Ir箔的EXAFS谱图进行FT变换后的径向分布函数(图10)显示,键长为2.5Å的Ir-M键只在SZIO中比较明显,而基本没有出现在SIO材料中;从EXAFS谱图计算得到的Ir-O配位数显示,在SZIO中,Ir-O配位数只有4.4,小于SIO中的6.0。以上证据皆表明SZIO材料中存在晶体缺陷(氧空位)。
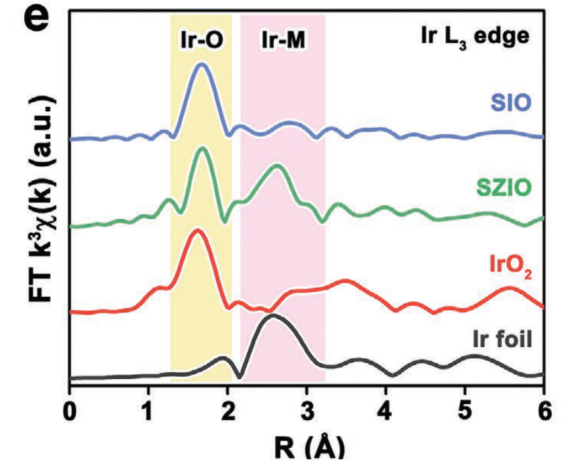
研究表明,合金化策略对于材料的体相与表面性能均产生了积极作用。相较于SIO,SZO的合金化作用减小了催化剂的粒径尺寸,并适当减弱了Ir位点原本对吸附氧中间体过强的吸附,从而有效提高了SZIO的催化活性。
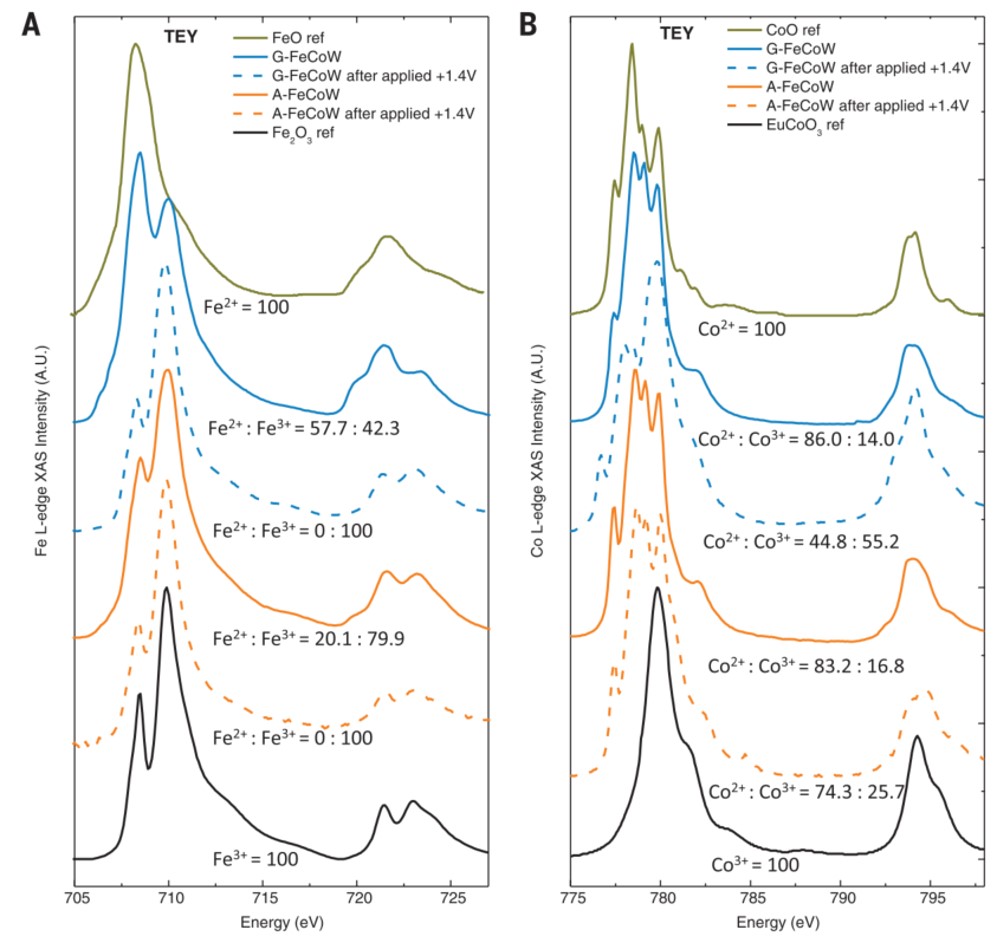
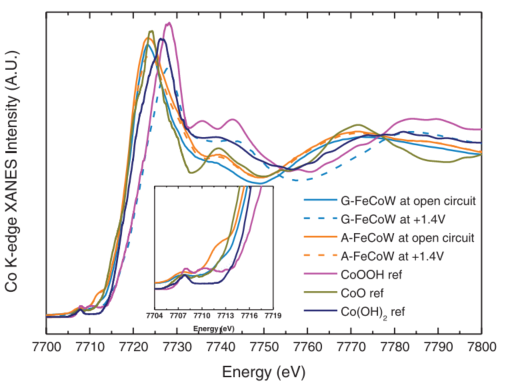
例6[9]:Bo Zhang等人通过理论计算得出W和Fe的双掺杂可提高CoOOH的OER性能,因此他们利用环氧丙烷溶胶凝胶法可控水解金属离子,并通过超临界CO2干燥,制备了原子级分散的FeCoW无定形氢氧化物(煅烧之后的材料记作A-FeCoW,未经煅烧的凝胶状材料记作G-FeCoW)并对其进行了XAFS表征。
由Fe和Co的TEY-XAS谱图(图11)可得,在1.4V的电位下,G-FeCoW中Fe会被氧化至+3价,但Co在A-FeCoW和G-FeCoW中的氧化还原行为不同。在A-FeCoW中,即使施加了1.4V的电位,A-FeCoW中Co2+的含量仍很高,而相比之下G-FeCoW在此电位下已基本转化为Co3+,这与Co K边XANES谱图(图12)的结果是吻合的。
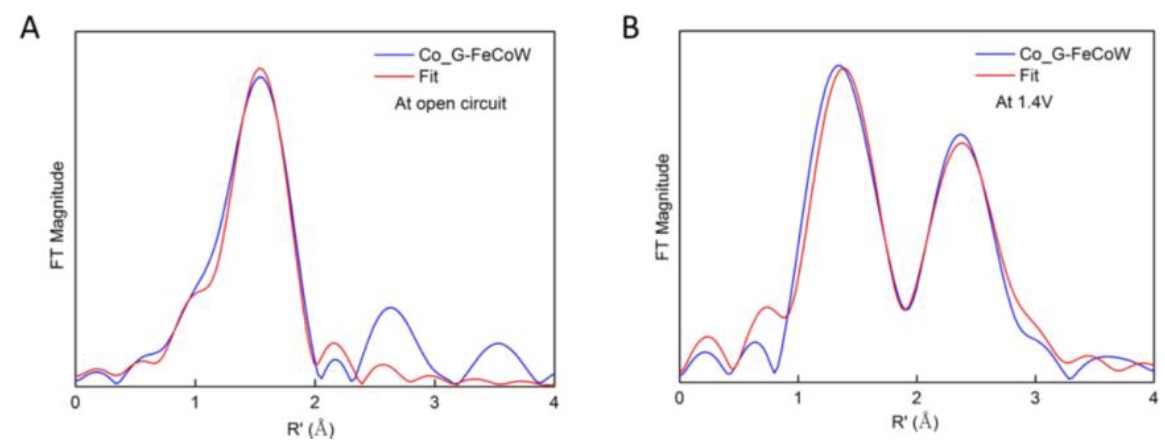
对Co K边EXAFS谱图进行FT变换,其径向分布函数(图13)表示,对G-FeCoW施加1.4V的电位后,Co-O键键长由2.06Å缩短为1.91Å,同样证明Co价态的升高。
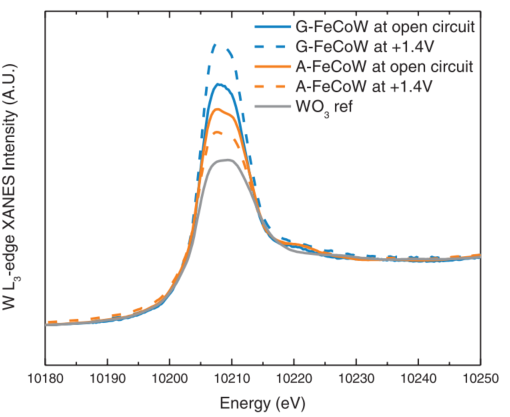
该课题组还对W的L3边XANES谱图(图14)进行了分析。由谱图可知,A-FeCoW在开路电压时的吸收峰较弱,这可归因为煅烧时失去结合水。当施加电位后,G-FeCoW的峰强明显提升,表示[WO6]八面体的畸变加强,这与DFT计算结果是吻合的。
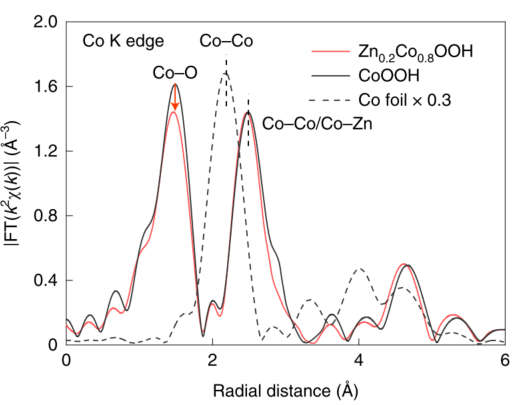
例7[10]:Zhen-Feng Huang等合成了一种ZnxCo1-xOOH材料,并通过XAFS研究其元素价态和配位情况。由Co K边EXAFS谱图变换得到的径向分布函数(图15)可得,在Zn0.2Co0.8OOH中键长2.41Å的Co-M峰与CoOOH相比基本无变化,说明Zn均匀取代了[CoO6]八面体中的Co3+位点,而键长1.48Å的Co-O键对应的峰强度出现了明显的下降,代表Co-O配位数的降低,这说明Zn取代后,材料中出现了更多氧空位。根据该发现,他们配合DFT理论计算提出了一种晶格氧(LOM)OER催化机理,解释了Zn0.2Co0.8OOH催化活性与pH有关的原因。
例8[11]:Ye Zhou等人将Fe掺杂入ZnCo2O4尖晶石结构中,并通过XANES研究了Zn、Fe、Co的价态。从Zn、Fe、Co的K边 XANES谱图(图16a,b)中可发现,随着Fe掺杂比例的升高,Zn的价态保持不变而Co的价态呈现先升高后降低的趋势,并在Fe:Co比达1:4时,Co价态达到最高值~3.4.后续的电化学测试中也证明Co的价态越高,催化性能越好。
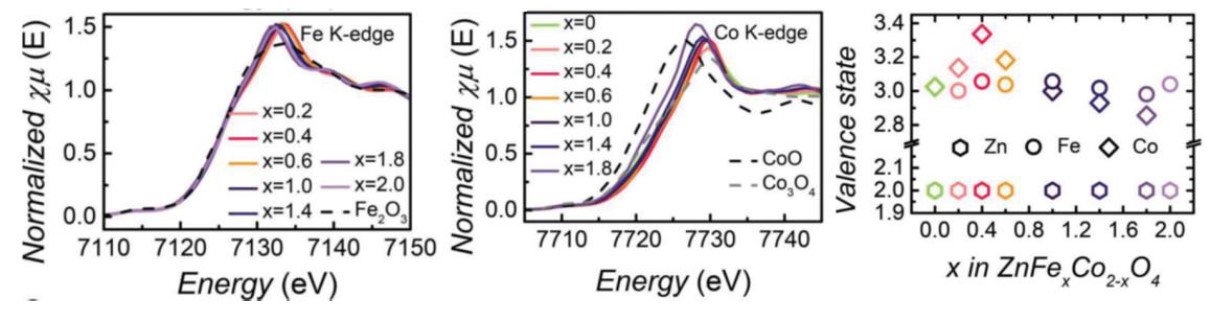
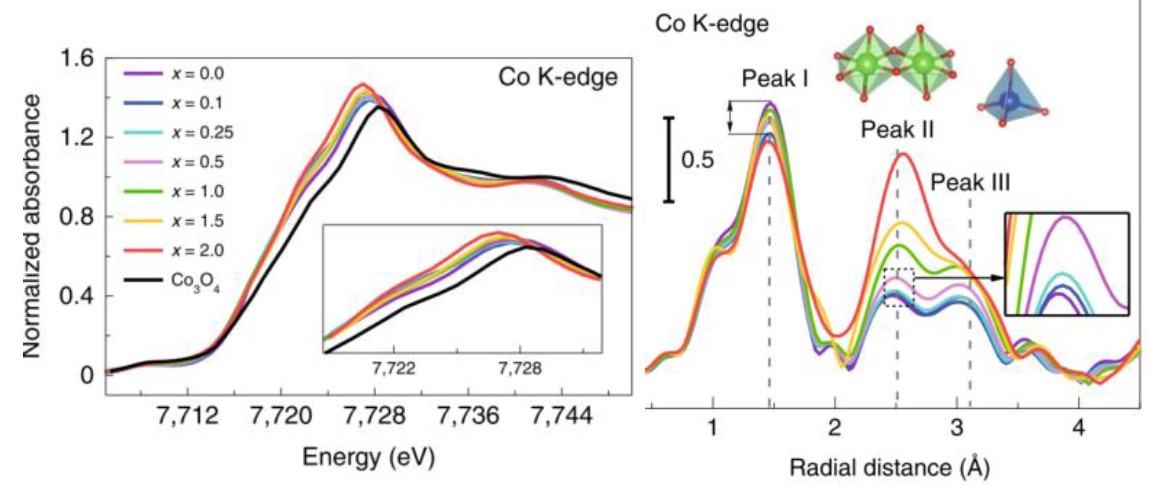
例9[12]:Tianze Wu等人将用Fe部分取代CoAl2O4中的Al,制备出高效的OER电催化剂(CoFe0.25Al1.75O4)。Fe的取代有助于在OER条件下将表面重构为高活性钴氢氧化物。Co K边XANES谱图(图17a)显示,尽管Fe3+的电负性强于Al3+,Fe的取代非但没有升高,反而发生了下降。研究人员进而用Co K边EXAFS谱图经FT变换的径向分布函数解释了这种反常现象的产生原因。由图17b可见,Co-O键对应的峰强略有下降,说明Co-O键的配位数有所降低,也表明有更多氧空位生成。
3.3 XAFS的前景与展望
XAFS技术是当前材料科学领域(尤其是表面化学和催化科学)最先进的技术之一。XAFS的发展加深了人类对材料的认识,比如复杂化学环境中原子、电子的相互作用。相信随着XAFS技术的继续进步(如第四代同步辐射光源、各种原位、飞秒级探测手段),会有更多重大科学成果出现。
参考文献
[1]寇元.固体催化剂的研究方法 第五章 XANES方法[J].石油化工,2000(09):712-722.
[2]寇元,邹鸣.固体催化剂的研究方法 第六章 EXAFS方法[J].石油化工,2000(10):802-811.
[3]H. Aritani, T. Tanaka, T. Funabiki, et al. Study of the Local Structure of Molybdenum−Magnesium Binary Oxides by Means of Mo L3-Edge XANES and UV−Vis Spectroscopy[J]. The Journal of Physical Chemistry, 1996, 100(50):19495-19501
[4]Z. Zhuang, Y. Li, Y. Li, et al. Atomically dispersed nonmagnetic electron traps improve oxygen reduction activity of perovskite oxides[J]. Energy & Environmental Science, 2021, 14(2):1016-1028
[5]J. Suntivich, W. T. Hong, Y.-L. Lee, et al. Estimating Hybridization of Transition Metal and Oxygen States in Perovskites from O K-edge X-ray Absorption Spectroscopy[J]. The Journal of Physical Chemistry C, 2014, 118(4):1856-1863
[6]Y. Tong, J. Wu, P. Chen, et al. Vibronic Superexchange in Double Perovskite Electrocatalyst for Efficient Electrocatalytic Oxygen Evolution[J]. J Am Chem Soc, 2018, 140(36):11165-11169
[7]G. Fu, W. Li, J. Y. Zhang, et al. Facilitating the Deprotonation of OH to O through Fe(4+) -Induced States in Perovskite LaNiO3 Enables a Fast Oxygen Evolution Reaction[J]. Small, 2021, e2006930
[8]X. Liang, L. Shi, R. Cao, et al. Perovskite-Type Solid Solution Nano-Electrocatalysts Enable Simultaneously Enhanced Activity and Stability for Oxygen Evolution[J]. Adv Mater, 2020, 32(34):e2001430
[9]B. Zhang, X. Zheng, O. Voznyy, et al. Homogeneously dispersed multimetal oxygen-evolving catalysts[J]. Science, 2016, 352(6283):333
[10]Z.-F. Huang, J. Song, Y. Du, et al. Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts[J]. Nature Energy, 2019, 4(4):329-338
[11]Y. Zhou, S. Sun, J. Song, et al. Enlarged CoO Covalency in Octahedral Sites Leading to Highly Efficient Spinel Oxides for Oxygen Evolution Reaction[J]. Adv Mater, 2018, 30(32):e1802912
[12]T. Wu, S. Sun, J. Song, et al. Iron-facilitated dynamic active-site generation on spinel CoAl2O4 with self-termination of surface reconstruction for water oxidation[J]. Nature Catalysis, 2019, 2(9):763-772